How to Extract Coding Sequence With Python Without Biopython
Bio.SeqIO module of Biopython provides a wide range of simple uniform interfaces to input and output the desired file formats. This file formats can only deal with the sequences as a SeqRecord object. Lowercase strings are used while specifying the file format. The same formats are also supported by the Bio.AlignIO module. The list of the file formats is given below :
| File Format | Description |
|---|---|
| abi | It is sequencing trace format for Applied Biosystem, reads the Sanger capillary sequence including PHRED quality scores for base calls. Each file contains only one sequence so no point of indexing the file. |
| abi-trim | Same as the abi format but has a quality trimming with Mott's algorithm. |
| ace | Reads the overlapping or contiguous sequences from the ACE assembly file. |
| cif-atom | Used to figure out the partial protein sequence by structure based on atomic coordinates |
| cif-seqres | Determines the complete protein sequence by reading a macromolecular Crystallographic Information File(also known as mmCIF) as defined by the _pdbx_poly_seq_scheme record. |
| embl | EMBL(Protein and DNA seq file format) flat file format, uses Bio.GenBank internally. |
| fasta | A generic sequence file format, each record starts with a line starting with > character followed by other sequence lines. |
| fasta-2line | Strict interpretation of FASTA file format by no line wrapping(i.e. using two line per record). |
| fastq | A FASTA variant with Sanger used to store PHRED sequence quality values with an ASCII of offset 33. |
| fastq-sanger | Alias for FASTA having consistency with BioPerl and EMBOSS |
| fastq-solexa | Original Solexa/Illumnia interpretation of FASTA format which is used to encode solexa quality scores with ASCII having offset of 64. |
| fastq-illumina | Solexa/Illumnia 1.3 to 1.7 version of FASTA format used to encode PHRED quality scores with ASCII of offset 64. |
| gck | Local format used by Gene Construction Kit. |
| genbank | GenBank or GenPept flat file format. |
| gb | Alias for genbank, having consistency with NCBI Entrez Utilities. |
| ig | IntelliGenetics file format, appears to be same as the MASE alignment format. |
| imgt | EMBL variant format from IMGT, where feature tables are intentionally allowed for longer feature types. |
| nib | UCSC nib file format for nucleotide uses 4-bits(1-nibble) to represent one nucleotide(2 nucleotides require i byte). |
| nexus | NEXUS multiple alignment format, also called PAUP format. |
| pdb-seqres | Reads PDB(Protein Data Bank) file and determines complete data sequence by header. |
| pdb-atom | Determines partial protein sequence by structure based on atomic coordinates section of file. |
| phd | These files are output of PHRED, used by PHRAP and CONSED for input. |
| pir | FASTA variant created by NBRF(National Biomedical Research Foundation) for Protein Information Resource database(PIR), presently art of UniProt. |
| seqxml | Simple XML file format. |
| sff | Standard Flow gram Format binary files created by Roche 454 and IonTorrent/IonProton sequencing machines. |
| sff-trim | Standard Flow gram Format (SFF) applies trimming to listed files. |
| snapgene | Local format used by SnapGene. |
| swiss | Plain text Swiss-Pro also called UniProt format. |
| tab | Simple two column tab separated sequence file, each line stores record identifier and sequence. |
| qual | FASTA variant having PHRED quality values from sequencing DNA. |
| uniprot-xml | UniProt XML format replacement of SwissProt plain text format. |
| xdna | DNA Strider's and SerialCloner's local format, used by Christian Marck. |
Below implementation explains about how to parse two of the most popular sequence file formats, i.e. FASTA and GenBank.
Attention geek! Strengthen your foundations with the Python Programming Foundation Course and learn the basics.
To begin with, your interview preparations Enhance your Data Structures concepts with the Python DS Course. And to begin with your Machine Learning Journey, join the Machine Learning - Basic Level Course
FASTA:
It is the most basic file format to store sequence data. Originally FASTA was a software package created during early evolution of Bioinformatics for sequence alignment of proteins and DNA, mostly used for searching similarities. Below is a simple example of parsing FASTA file format:
Example:
To get the input file used click here.
Python3
from Bio.SeqIO import parse
file = open ( 'is_orchid.fasta' )
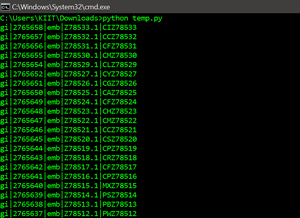
for record in parse( file , "fasta" ):
print (record. id )
Output:
GeneBank:
Richer sequence format for genes which includes various annotations. Parsing the GenBank format is as simple as changing the format option in Biopython parse method. Below is a simple example of parsing GenBank file format:
Example:
To get the input file used click here.
Python3
from Bio import SeqIO
from Bio.SeqIO import parse
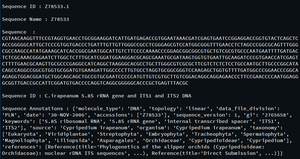
seq_record = next (parse( open ( 'is_orchid.gbk' ), 'genbank' ))
print ( "\nSequence ID :" , seq_record. id )
print ( "\nSequence Name :" , seq_record.name)
print ( "\nSequence :" , seq_record.seq)
print ( "\nSequence ID :" , seq_record.description)
print ( "\nSequence Annotations :" , seq_record.annotations)
Output:
How to Extract Coding Sequence With Python Without Biopython
Source: https://www.geeksforgeeks.org/biopython-sequence-file-formats/
0 Response to "How to Extract Coding Sequence With Python Without Biopython"
Post a Comment